
.
Elles sont au nombre de 7000 (répertoriées), on en dépiste à présent en routine plus de 200 au niveau du génome des parents, avant ou durant la grossesse (si notion de risque familial). On rencontre ces affections plus souvent chez les isolats génétiques (cousins germains, Amiches, vallée de montagnes, îles lointaines, Roms, etc…)
La future mère est porteuse du gène … pas de risque si ton époux n’a pas le même défaut (fragilité qui est quand même rare). Si les deux parents ont ce défaut :
.
Le mécanisme est toujours le même : déficit d’une enzyme qui conduit à l’accumulation du précurseur (toxicité – cas de la maladie de Gaucher) et au déficit de la molécule finie (cas de la mucoviscidose).
Précurseur A …> enzyme déficient …> Produit fini B (actif)
.
Il y a de nombreuses recherches en cours sur ce type d’affections. Trois types de traitements peuvent être proposés :
.
— Troubles des acides aminés : la fonction amine (NH)
.
1/ La Phénylcétonurie
Deux formes : minime et maligne.
La forme bénigne persistante est à un gène, donc pas besoin de traitement. Mais la grossesse d’une personne porteuse constitue un risque, car cette augmentation de la phénylalanine va donner naissance à un enfant pas forcément porteur mais avec cardiopathie, microcéphalie (retard mental associé) et retard mental. C’est à dépister avant la grossesse et pendant celle-ci, on donne un régime pour la substance. Les enfants porteurs naissent cliniquement normaux mais peuvent, dès les 1ers mois, avoir un retard psychomoteur, un trouble de la pigmentation (peau, cheveux et yeux plus clairs), être irritable, hyperactif, présenter des spasmes en flexion, des troubles du tonus musculaire avec mouvements involontaires, pouvant aller jusqu’aux crises convulsives .
L’urine des enfants est riche de cet acide aminé, avec odeur de nid de souris. On dépiste à la maternité le bébé, et s’il est positif, il sera normalisé par un régime alimentaire (lait spéciaux pauvre en phénylalanine). ).
.
2 – L’Homocystinurie
L’aspect clinique est le même. Manifestations constantes : retard mental, biotype marfanoïde (longues extrémités et de stature élevée avec risque de mourir de problèmes cardiaques (par fragilité du tissu conjonctif, touchant la crosse de l’aorte qui peut éclater). Augmentation du tonus musculaire, convulsions et troubles. Certaines formes peuvent présenter des thrombus veineux et des maladies emboliques provoquant hémiplégie ou autres syndromes neurologiques, anémie mégaloblastique (tableau d’hypoxie). On réduit donc les apports en méthionine et on administre des vitamines C et B6.
.
3 – Hyperglycémie sans cétose
.
4 – Maladie du sirop d’érable : urine sente le sirop d’érable.
.
Métabolisme des acides organiques :
.
Troubles du cycle de l’urée :
.
Mucopolysaccharidoses (MPS)
Sucres complexes accumulés dans l’appareil digestif des cellules qui n’est plus dégradé correctement. Ce qui induit des troubles divers comme : l’opacification de la cornée ++, une cyphose thoraco-lombaire, épaississement des poignets et bras, augmentation du volume du foie, petite taille, gros crâne, abdomen protubérant, troubles multiples de l’ossification, retard mental.
Pas de symptômes à la naissance, mais les troubles apparaissent progressivement après quelques mois avec un tableau spécifique en fonction de la forme déduisant la forme enzymatique. L’étude radiologique permet aussi de voir les atteinte de la colonne vertébrale (surtout transition thoraco-lombaire), crâne (surtout selle turcique), mains et pieds. On a essayé de faire des greffes de moelles mais c’est expérimental encore.
On distingue actuellement :
.
Sphingolipidoses
Grosses molécules grasses saturées qui s’accumulent dans les tissus.
.
* La maladie de Nieman-Pick
Les formes A, B, C, se trouvent dans le cerveau et organes lymphoïdes (rate, ganglions de l’intestin).
.
* La maladie de Gaucher se présente sous trois formes cliniques :
1/ le type 1, dit « sans atteinte du système nerveux », mais avec des manifestations hématologiques (anémie, thrombocytopénie), hépatique (hépatospénomégalie) ou osseuses (fractures). L’âge du début de la maladie et sa gravité sont variables, pouvant même être asymptomatique. Cette forme bénéficie d’un traitement veineux par l’enzyme manquant : bons résultats (mais produits chers : ZAVESCA).
2/ le type 2, dit « forme aiguë neuropathique », est également très hétérogène : début précoce ou tardif, hépatomégalie, troubles du mouvement des globes oculaires, du tonus, mouvements involontaires … selon le cas.
3/ le type 3, dit « forme subaiguë neuropathique », présente une hépato- splénomégalie, des lésions osseuses et occasionnellement d’autres organes.
.
* La maladie de Fabry, ou syndrome de Ruiter-Pompen-Wyers est une maladie génétique, liée au chromosome X, résultant d’un déficit enzymatique de l’alpha-galactosidase lysosomale avec accumulation de globotriaosylcéramide et de digalactosylceramide dans les cellules.
C’est une maladie rare, bien que probablement sous évaluée (moins de 1 naissance pour 50 000 garçons). Les femmes porteuses du gène muté peuvent en présenter des signes, mais plus tardifs et souvent moins graves. La forme classique survient chez le jeune garçon lorsque l’activité enzymatique est inférieure à 1%.
Les premiers signes commencent dans l’enfance ou l’adolescence par des crises douloureuses des extrémités (acroparesthésies), souvent très gênantes, l’apparition de lésions vasculaires cutanées (angiokératomes se situant essentiellement sur le torse ou en péri-ombilical), une baisse de la sécrétion de sueur, des anomalies de la cornée (mais sans conséquence sur la vision), une cataracte et la présence d’une protéinurie d’apparition plus tardive. Une insuffisance rénale s’installe dans près de la moitié des cas. Les symptômes digestifs sont également fréquents, comportant nausées, douleurs abdominales, diarrhée…
Malgré un traitement bien conduit, la majorité des hommes auront des complications cardiovasculaires (essentiellement une hypertrophie ventriculaire gauche et des accidents vasculaires cérébraux). Cette atteinte cardiaque peut se traduire par des troubles du rythme ventriculaire avec risque de mort subite.
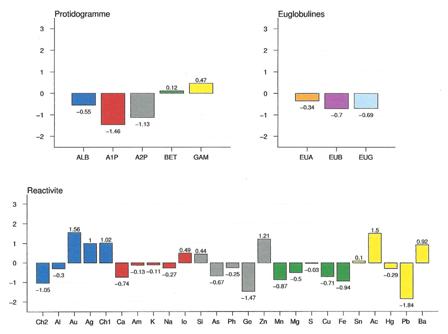
Les femmes porteuses hétérozygotes auront une vie quasiment normale avec quelques manifestations au fur et à mesure de leur vieillissement. Dans le cas présenté ci-dessous, il s’agit d’une femme jeune, dont le seul symptôme gênant est une rosacée :
.
On remarquera l’aspect de sécheresse (hypo Albumines / Alpha 1 / Euglobulines) et la dissociation des tests du pôle rénal (en bleu) de ce BNS 24.
.
* Maladie de Krabbe
Maladie autosomique récessives causée par un déficit enzymatique (cerebrosite sulfatase) : matériel lipidique intra-accumulé qui colore les tissus en marrons. IRM : démyélinisation de l’encéphale. Intérêt à cause de la possibilité de faire une thérapie génique (cf. téléthon).
.
Désordre du métabolisme du cuivre
.
Désordre du métabolisme des purines : syndrome de Lesch-Nyhan
Affection liée au chromosome X, comme l’hémophilie et le daltonisme !
.
En construction … tant le sujet est vaste !