
.
Groupe d’affections graves et évolutives, caractérisés par la présence d’AC anti-cellules endothéliales (ANCA). Des vascularites sans ANCA se rencontrent dans le cadre des maladies auto-immunes systémiques (ex.: LED). On peut distinguer :
.
.
La maladie de HORTON (décrite en 1932), artérite temporale giganto-cellulaire des sujets âgés (HLA DR4 ou DR7). La prévalence est d’environ 1 sur 1000 pour les 60-69 ans et va jusqu’à 1 sur 120 chez les plus de 80 ans.
.
.
Diagnostic différentiel : l’algie vasculaire de la face et la névralgie du trijumeau.
.
A noter le risque de cécité bilatérale qu’une corticothérapie précoce peut prévenir, de nécrose du cuir chevelu (rare) ou de la langue (rarissime). Le traitement corticoïde doit être poursuivi pendant environ deux ans, les rechutes étant fréquentes.
Un BNS est indispensable pour le choix d’un traitement adapté.
.
L’artérite de Takayatsu (décrite en 1908), aortite des femmes jeunes (grave, mais assez rare) avec troubles vasculaires ischémiques (présence d’un AC anti-aorte).
.
La poly-artérite noueuse (PAN – décrite dès 1866) qui est souvent associée à une infection chronique à HBV (30% des malades) ou HIV. Traitement classique par corticothérapie et immunodépresseurs. C’est une vascularite nécrosante (HLA B8) et inflammatoire segmentaire des artères de petit et de moyen calibre, de symptomatologie polymorphe par retentissemant sur la fonction des organes irrigués par ces vaisseaux. Le diagnostic est assuré par la biopsie cutanée.
Un tel tableau clinique nécessite un bilan BNS (à refaire tous les six mois) qui fera le point des troubles des volumes corporels et des régulations. Un traitement personnalisé pourra être alors choisi et changer le cours de la maladie.
Si vous souhaitez réaliser un BNS12 ou 24, cliquez sur ce lien : www.mybiobox.com
.
.
Les cryoglobulinémies polyclonales à dépôts d’IgM, dont les symptômes s’apparentent au syndrome de Raynaud : Secale cornutum (pb), Plumbum, Arnica (ecchymoses), Lachesis, Carbo vegetabilis (cyanose des extrémités), Gentiana lutea (k).
.
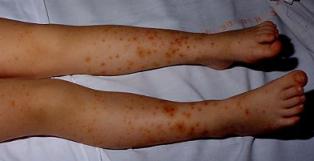
Le purpura rhumatoïde, aux dépôts d’IgA et de C3 sur la peau, le rein, le tube digestif et les articulations. Purpura pétéchial pouvant confluer en plaques ecchymotiques, parfois associé à des macules, des papules, des plaques urticariennes, des œdèmes localisés (cheville), de type orthostatique : prédominant aux membres inférieurs, pouvant toucher les coudes et les avants bras, rare sur le tronc et la face. Evoluant par poussées, souvent déclenchées par la reprise de l’activité.
.
.
Il peut s’accompagner d’arthralgies des grosses articulations, pouvant précéder, accompagner ou suivre l’atteinte cutanée, résolutive en quelques jours. Douleurs souvent intenses à type de coliques, avec vomissements, mais état général conservé (photo ci dessus).
.
Organothérapie/sérothérapie : foie et rate (SRE)
.
Le syndrome des anti-phospholipides (SAPL) est une maladie emboligène à rechute, diagnostiquée par le dosage des AC anti-cardiolipines et anti B2-glycoprotéine 1. Traitement prophylactique aux AVK (ex.: Sintron). Cf. BNS ci-dessous :
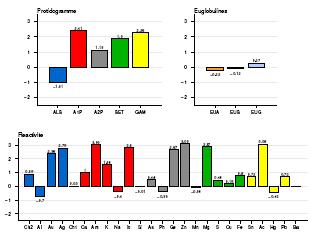
Manifestations vasculaires :
Manifestations obstétricales : Boffa et al. 1995, Silver et al. 1992, proposent comme critères du diagnostic du syndrome des anticorps antiphospholipides : hématome rétroplacentaire, pré-éclampsie, éclampsie, HELLP syndrome et les accidents ischémique foetaux.
Autres manifestations peuvent être inclues :
Les récidives sont fréquentes. Curieusement, un épisode thrombotique artériel initial tente à récidiver dans un autre territoire artériel et il en est de même pour un événement veineux. La fréquence des récidives a été estimée à environ 10 % à 4 ans.
.
La maladie de Wegener est une vascularite avec des granulomes dans le nez, les sinus, les oreilles et les poumons. La maladie est donc aussi appelée « granulomatose de Wegener ». Cependant, tous les organes peuvent être atteints, notamment les yeux, le cœur, la peau, les articulations, les reins et le système nerveux.
.
Sont également à classer dans ce groupe :
.
Le syndrome de Goodpasture
Affection rénale (polyurie, polydipsie, protéinurie, hématurie microscopique), accompagnée de :
dont l’évolution habituelle se fait vers l’insuffisance rénale terminale.
.
La maladie de Schönlein-Henoch
La Pan-angéite microscopique
La maladie de Chug et Strauss
.
3/ Les vascularites à prédominance veineuse :
.
La Maladie de La Peyronie (développée sur ce site au service « Uro-génital »)
.
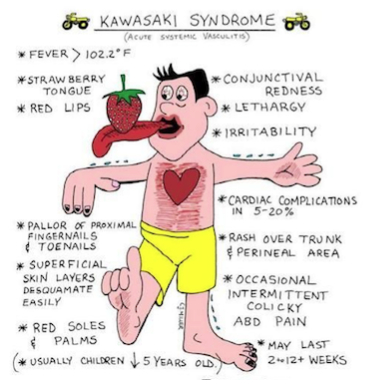
La Maladie de Kawasaki (décrite en 1974) qui touche parfois les enfants, avec fièvre, rash diffus, conjonctivite et adénopathies.
.
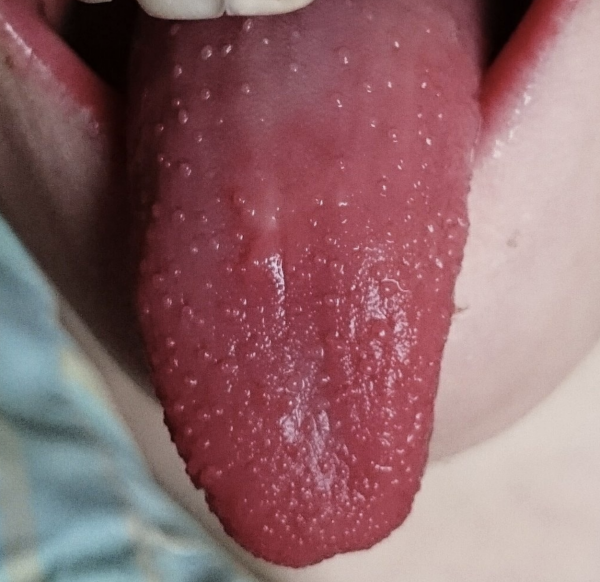
Langue « framboisée …
.
L’Angéïte leucocytoclastique
Le BNS apporteront – bien sûr – des éléments d’évaluation essentiels, ainsi que des propositions thérapeutiques parfois brillantes (stabilisation fréquente des PAN et des purpuras rhumatoïdes).
.
Ces affections pourront bénéficier de remèdes homéopathiques complexes à polarité vasculaire : AESCULUS Comp., ANGIO-INJEEL, ARSURANEEL, ARTERIA-Heel, CIRCULO-Heel, SECALE Comp., TRAUMEEL … toujours, bien sûr, avec : PLACENTA Comp. et UBICHINON Comp.
Avec les sels de terrain : EFFICOMPLEX n°5 et 6 alternés (pharmacie des Bergues – Genève).
.
Pour plus de détails techniques : Groupe Français d’Etude des Vascularites (GFEV) : http://www.vascularites.org/